How to use SDM
Main page

About SDM
SDM is a computational method to predict the change in protein stability upon the introduction
of a single mutation. It is a knowledge-based approach that uses conformationally constrained environment-dependent amino acid substitution tables to predict the change the protein stability between wildtype and mutant protein.
Submission page

How to run a prediction
To run a prediction:
(1) Click on "Predict" to open the submission page.
(2) Provide the structure of the wild-type protein,
which must comply with the PDB format.
(3) A single mutation or a file with a list of mutations to be analysed should be provided.
For single mutation mode, a mutation code consists of wild-type code, residue position and mutant code (using the one letter amino acid code as shown in example). Similar format is used to list mutations in a file (An an example mutant list file could be downloaded by clicking the like "2OCJ_mut_list". A maximumm of 20 mutations are allowed in the mutant list file.
Residue position must be consistent with the PDB file. The chain ID must also be provided for both single muation and mutation list option.
(4) Check this option to predict reverse mutation.
(5) You are then ready to submit your query for analysis.
Results page
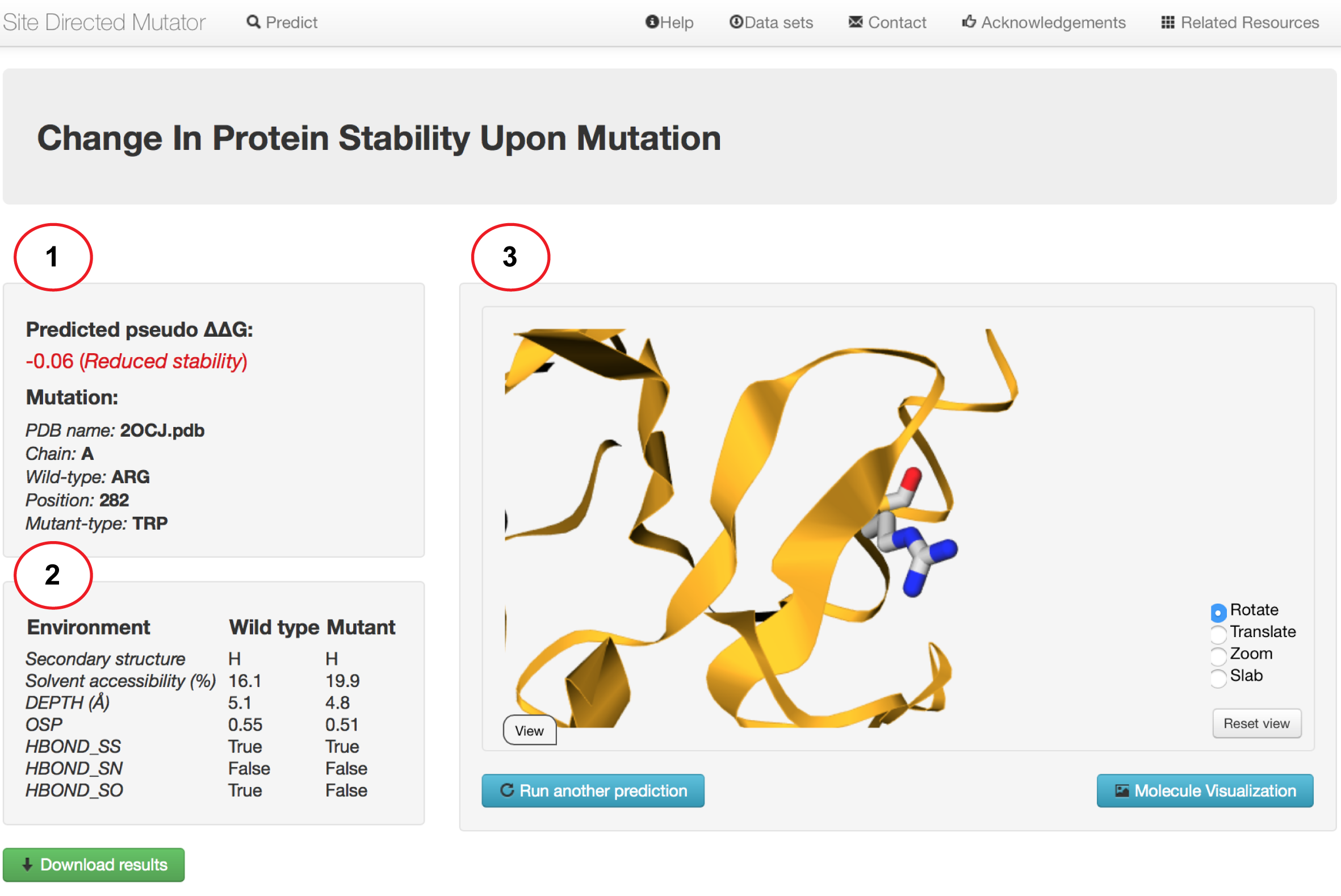
Results - Single mutation
(1) Your results for a single mutation will be displayed once calculations are completed. The results will display the predicted
change in protein stability upon mutation (pseudo ΔΔG). A negative (in red) and positive (in blue) value corresponds
to mutation predicted to be destabilising and stabilising respectively. Complementary information displayed include:
(2) A summary of the mutation is presented highlighting the wild-type residue, position
number, chain and the mutant residue. A separate panel list the structural features including the main chain conformation class of the secondary structure, solvent accessibility, residue depth, residue occuluded surface packing (OSP), sidechain-sidechain hydrogen bond (HBOND_SS), sidechain-main chain amide hydrogen bond (HBOND_SN), sidechain-main chain carbonyl hydrogen bond (HBOND_SO) for the wildtype and mutant residue.
(3) The protein structure with its wildtype residue environment can be visualised directly from the server.
Results page

Results - List of Mutations
(1) Your results for a list of mutations will be displayed in a table format with the following information:
Mutation field contain the information about the mutation including the wildtype single letter amino acid code followed by the mutation residue position and the mutant amino acid code.
The main chain conformation class of the secondary structure for the wildtype (WT_SSE) and mutant (MT_SSE) residue.
Residue percentage relative solvent side chain accessibility (RSA) for wildtype (WT_RSA %) and mutant (MT_RSA %).
Residue depth for wildtype (WT_DEPTH) and mutant (MT_DEPTH) in angstrom units.
Residue occluded surface packing for wildtype (WT_OSP) and mutant (MT_OSP).
Sidechain-sidechain hydrogen bond (HBOND_SS), sidechain-main chain amide hydrogen bond (HBOND_SN) and sidechain-main chain carbonyl hydrogen bond (HBOND_SO) for the wildtype and mutant residue.
Contact page
Getting in touch
In case you experience any trouble using SDM or have any suggestions or comments, please do not hesitate in contacting us either via e-mail or through the online form.